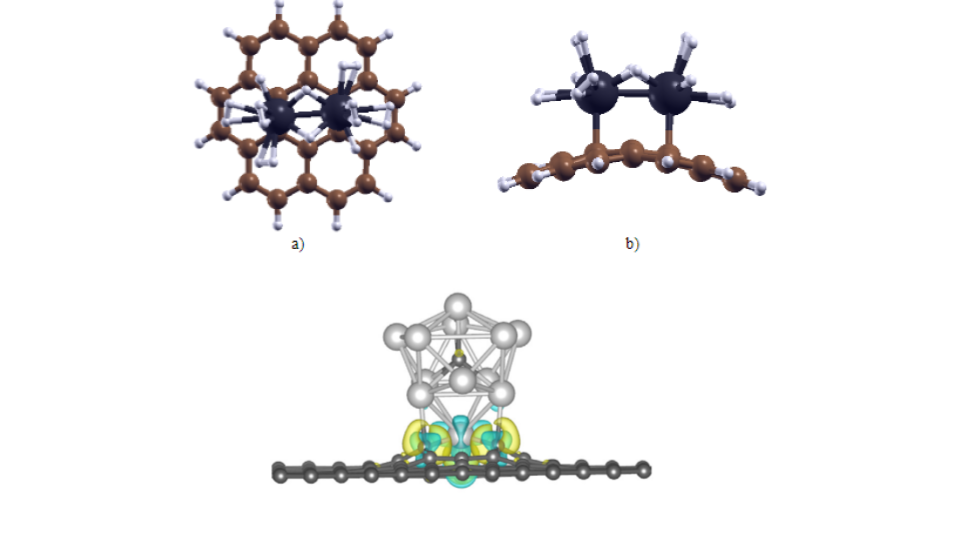
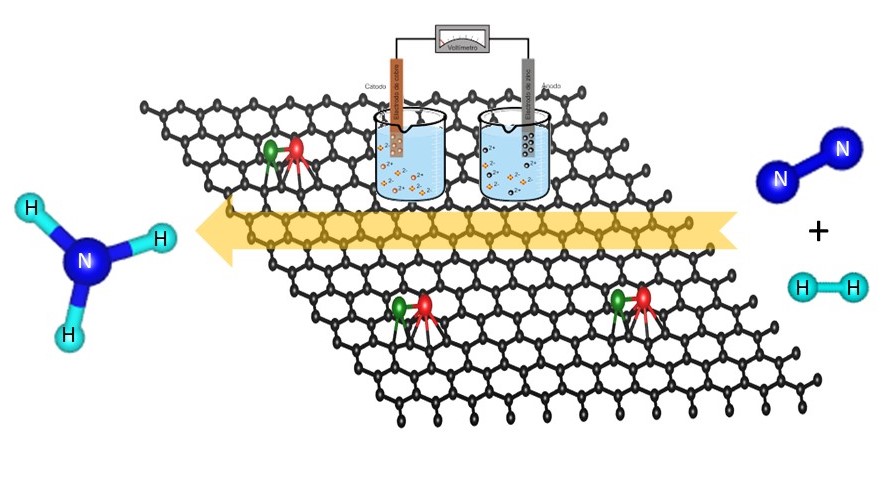
N2 fixation and activation is essential for the sustenance of life. N2 though being the most abundant constituent in the air, its fixation and activation is a high energy process on account of its triple bond making it a nearly inert gas. Bosch-Haber process, the well known chemical transformation method from N2 to NH3 is the main industrial procedure for production of ammonia in the world and consumes 2% of world's energy. In this context, the research in this group is to identify cost-effective catalysts for N2 activation and its dissociation using computational methods such as Density Functional Theory (DFT) and hybrid methods such as QM/MM. The catalysts computationally studied in the group range from transition metal based organic ones to organic-inorganic hybrid catalysts as represented below.
CO2, a major green house gas is the distrubing the carban balance within our atomosphere. In order to meet the ambitious Paris agreement, CO2, conversion to value added products, we need an accelerated design of efficient catalysts. Our research emphasis is to computationally design graphene based catalysts for effective conversion of CO2 to formic acid. The theoretical calculations (DFT) are followed by experimental studies to envisage a practical catalyst for the same. Represented below is one suh study where we have first computationally designed a catalysts and synthesized it to have formic acid at low overlay potentials.
Understanding the underlying electronic factors that contribute to the enhancement of the reactivity of Oxygen Reduction Reaction (ORR) and Hydrogen Evolution Reaction (HER) catalysts is an important research activity of the group. The theoretical inputs obtained from DFT studies are dynamically taken to the collaborating experimental researchers so as to envisage practically the better catalysts. There is plenty of scope to do both experimental and theoretical research work in this research topic. Visit the webpage of our experimental collaborator academic.ncl.res.in/profile/k.selvaraj
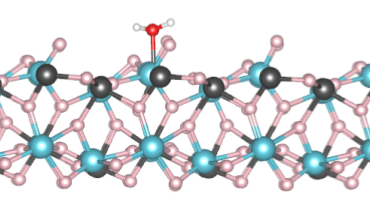
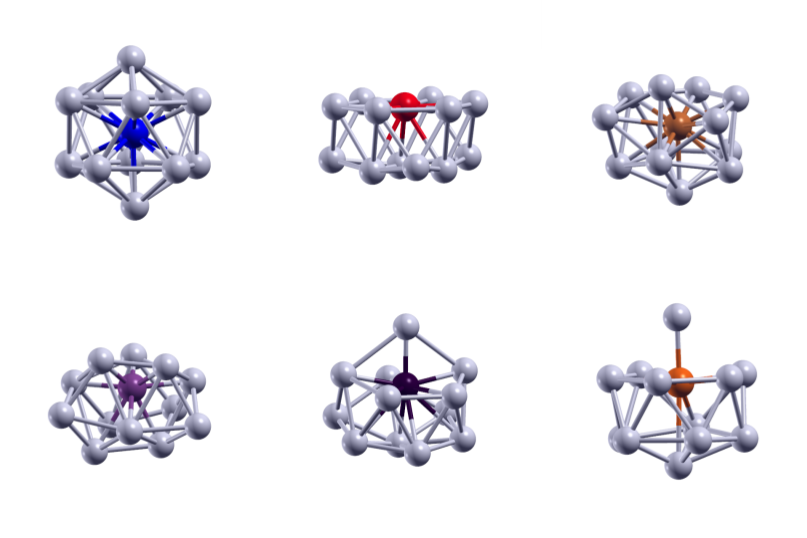
DFT, QM/MM and AIMD studies are applied to correlate the complexities of the structure-property relationship in atomic sized clusters.These studies help us build nanoparticles from a bottom up approach where we we have have an uniquely tuned property for a sized system.
Very little is understood about the delicate balance between the intra and inter molecular forces in macro-bio molecules such as lipds. We discern these relationships using state of art Density Functional Theory Methods and ab initio Molecular Dynamics (AIMD). This is a very exciting and yet fundamental work that brings insight on the way these molecules organize within theselves.
The extensive use of toxic chemicals like pecticides in agriculture pose a great threat to enviournment and mankind. Also the offloading of harmful waste in water bodies make our water supplies unfit for consumpion. Sensing and detection of toxic molecules like by 2D materials is based on adsorption properties of the later towards the former. Density Functional Theory (DFT) based calculations are applied to account for the adsorption capabiliites of graphene and graphene based materials towards of toxic materials. This is another work that is done in tandem with the experimentalists.
Morphological transformation of materials such as stripping of graphene nanotubes to ribbons, stiching of graphene sheets into tubes, transformation of fullerenes into graphene nanoflakes is an exciting area of research where there is plenty of scope to play with the chemical composition and morphology of novel 2D materials. Imrpoved catalysts for several applications can be designed using modeling and simulation tools such as Density Functional Theory (DFT) based methods and ab initio Molecular Dynamics (AIMD) based methods. This area is one of the on going research topics in the group.
Corrosion is an irreversible electrochemical process of industrially applied surfaces causing tremendous economic loss, environmental harm and wastage. Traditional corrosion inhibitors use environmentally harmful molecules that are designed by a trial and error approach. The systematic search for green and environmental friendly corrosion inhibiting molecules (green corrosion inhibitors) and their final applicability as corrosion materials is critical, especially in the light of increasingly harsh environmental conditions. The search for these inhibitors can be accelerated by an effective multi-scale computational approach. This project is a collaborative one with several theoretical chemists in NCL participating towards computational design of novel corrosion inhibitors. Modeling the physico-chemical properties of various inhibitors will be done using different computational methodologies such as DFT, atomistic and mesoscopic methods (http://academic.ncl.res.in/profile/d.sengupta), and analytical methods (http://academic.ncl.res.in/profile/mb.sarika).
Efficient and low cost storage of gas molecules at near ambient temperature and pressure is very important. Gases like nitrogen and carbon dioxide are important precursors for various commercial chemical compounds. Efficient storage of hydrogen is necessary to realize the goal of the hydrogen economy. Thus this research aims at exploring and designing novel materials like 2D materials, atomic clusters, quantum confined nanomaterials, etc. Quantum chemical methods Density Functional Theory(DFT) and Ab Initio Molecular Dynamics(AIMD) methods are employed to study the various structural, electronic and structural properties of these materials.
|
|
.gif)

.png)